

« Back to all publications
Download this list in a RIS file or a BIB file or a PDF file
|
 |
|||||||
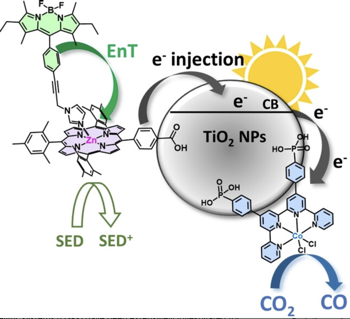
Dye-sensitized photocatalytic systems (DSPs) have been extensively investigated for solar-driven hydrogen (H2) evolution. However, their application in carbon dioxide (CO2) reduction remains limited. Furthermore, current solar-driven CO2-to-CO DSPs typically employ rhenium complexes as catalysts. In this study, we have developed DSPs that incorporate noble metal-free components, specifically a zinc-porphyrin as photosensitizer (PS) and a cobalt-quaterpyridine as catalyst (CAT). Taking a significant stride forward, we have achieved an antenna effect for the first time in CO2-to-CO DSPs by introducing a Bodipy as an additional chromophore to enhance light harvesting efficiency. The energy transfer from Bodipy to zinc porphyrin resulted in remarkable stability (turn over number (TON)=759 vs. CAT), and high CO evolution activity (42?mmol?g?1?h?1 vs. CAT). | ||||||||



 |
|
|||||||
So far, symmetry-breaking charge separation (SB-CS) has been observed with a limited number of chromophores and is usually inhibited by the formation of an excimer. We show here that thanks to fine-tuning of the interchromophore coupling via structural control, SB-CS can be operative with pyrene, despite its high propensity to form an excimer. This is realized with a bichromophoric system consisting of two pyrenes attached to a crown ether macrocycle, which can bind cations of different sizes. By combining stationary and time-resolved spectroscopy together with molecular dynamics simulations, we demonstrate that the excited-state dynamics can be totally changed depending on the binding cation. Whereas strong coupling leads to rapid excimer formation, too weak coupling results in noninteracting chromophores. However, intermediate coupling, achieved upon binding of Mg2+, allows for SB-CS to be operative. | ||||||||
|
 |
|||||||
There is a growing interest in developing dye-sensitized photocatalytic systems (DSPs) to produce molecular hydrogen (H2) as alternative energy source. To improve the sustainability of this technology, we replaced the sacrificial electron donor (SED), typically an expensive and polluting chemical, with an alcohol oxidation catalyst. This study demonstrates the first dye-sensitized system using a diketopyrrolopyrrole dye covalently linked to 2,2,6,6-tetramethyl-1-piperidine-N-oxyl (TEMPO) based catalyst for simultaneous H2 evolution and alcohol-to-aldehyde transformation operating in water with visible irradiation. | ||||||||
 |
|
|||||||
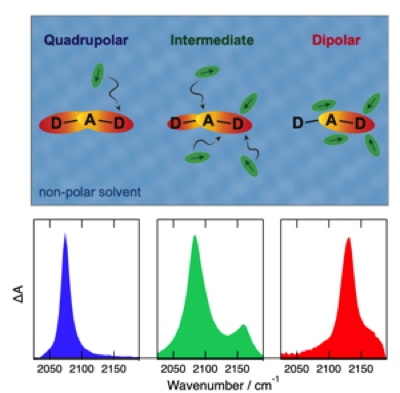
A large number of multipolar dyes undergo excited-state symmetry breaking (ESSB) in polar media. During this process, electronic excitation, initially distributed evenly over the molecule, localizes, at least partially, on one donorâââ‰â¬Åacceptor branch. To resolve its initial stage, ESSB is investigated with a donorâââ‰â¬Åacceptorâââ‰â¬Ådonor dye in binary mixtures of nonpolar and polar solvents using time-resolved infrared absorption spectroscopy. The presence of a few polar molecules around the dye is sufficient to initiate ESSB. Although the extent of asymmetry in a mixture is close to that in a pure solvent of similar polarity, the dynamics are slower and involve translational diffusion. However, preferential solvation in the mixtures leads to a larger local polarity. Furthermore, inhomogeneous broadening of the S1 <- S0 absorption band of the dye is observed in the mixtures, allowing for a photoselection of solutes with different local environments and ESSB dynamics. | ||||||||
|
 |
|||||||
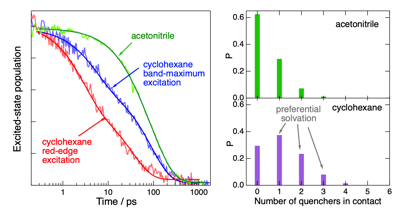
Electron transfer (ET) quenching in nonpolar media is not as well understood as in polar environments. Here, we investigate the effect of dipoleââ¬âdipole interactions between the reactants using ultrafast broadband electronic spectroscopy combined with molecular dynamics simulations. We find that the quenching of the S1 state of two polar dyes, coumarin 152a and Nile red, by the polar N,N-dimethylaniline (DMA) in cyclohexane is faster by a factor up to 3 when exciting on the red edge rather than at the maximum of their S1 ââ S0 absorption band. This originates from the inhomogeneous broadening of the band due to a distribution of the number of quencher molecules around the dyes. As a consequence, red-edge excitation photoselects dyes in a DMA-rich environment. Such broadening is not present in acetonitrile, and no excitation wavelength dependence of the ET dynamics is observed. The quenching of both dyes is markedly faster in nonpolar than polar solvents, independently of the excitation wavelength. According to molecular dynamics simulations, this is due to the preferential solvation of the dyes by DMA in cyclohexane. The opposite preferential solvation is predicted in acetonitrile. Consequently, close contact between the reactants in acetonitrile requires partial desolvation. By contrast, the recombination of the quenching product is slower in nonpolar than in polar solvents and exhibits much smaller dependence, if any, on the excitation wavelength. | ||||||||
 |
|
|||||||
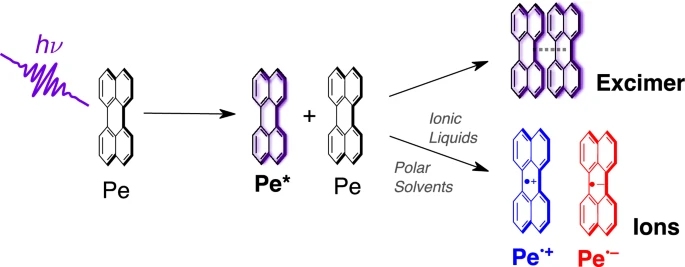
Photoinduced symmetry-breaking charge separation (SB-CS) results in the generation of charge carriers through electron transfer between two identical molecules, after photoexcitation of one of them. It is usually studied in systems where the two reacting moieties are covalently linked. Examples of photoinduced bimolecular SB-CS with organic molecules yielding free ions remain scarce due to solubility or aggregation issues at the high concentrations needed to study this diffusion-assisted process. Here we investigate the excited-state dynamics of perylene (Pe) at high concentrations in solvents of varying polarity. Transient absorption spectroscopy on the subnanosecond to microsecond timescales reveal that self-quenching of Pe in the lowest singlet excited state leads to excimer formation in all solvents used. Additionally, bimolecular SB-CS, resulting in the generation of free ions, occurs concurrently to excimer formation in polar media, with a relative efficiency that increases with the polarity of the solvent. Moreover, we show that SB-CS is most efficient in room-temperature ionic liquids due to a charge-shielding effect leading to a larger escape of ions and due to the high viscosity that disfavours excimer formation. | ||||||||
|
||||||||
Understanding the origin of electron-phonon coupling in lead halide perovskites is key to interpreting and leveraging their optical and electronic properties. Here we show that photoexcitation drives a reduction of the lead-halide-lead bond angles, a result of deformation potential coupling to low-energy optical phonons. We accomplish this by performing femtosecond-resolved, optical-pump-electron-diffraction-probe measurements to quantify the lattice reorganization occurring as a result of photoexcitation in nanocrystals of FAPbBr(3). Our results indicate a stronger coupling in FAPbBr(3) than CsPbBr(3). We attribute the enhanced coupling in FAPbBr(3) to its disordered crystal structure, which persists down to cryogenic temperatures. We find the reorganizations induced by each exciton in a multi-excitonic state constructively interfere, giving rise to a coupling strength that scales quadratically with the exciton number. This superlinear scaling induces phonon-mediated attractive interactions between excitations in lead halide perovskites. | ||||||||
Download this list in a RIS file or a BIB file or a PDF file
Last modified March 26 2024
Contact:
Eric Vauthey
Physical Chemistry Department - Sciences II - University of Geneva
30, Quai Ernest Ansermet - CH-1211 Geneva 4 (Switzerland)
© All rights reserved by Eric Vauthey and the University of Geneva
Design and code by Guillaume Duvanel